The survey conducted on notified bodies last year highlighted a critical issue impacting MDR and IVDR conformity assessment processes: an insufficient number of notified bodies certified to both regulations. This challenge was compounded by a concerning number of MDR and IVDR certified manufacturers, especially considering that most MDD and IVDD certificates would become void in 2024.
Towards the end of 2022 and the beginning of 2023, several MDCG guidance were issued. Finally, the European Commission published Regulation 2023/607, amending the MDR and IVDR by extending the transitional periods of medical devices and removing the “sell-off” deadline for both medical devices and IVDs.
The extension of transitional period undeniably provided manufacturers with more time to continue producing legacy devices while transitioning their qualify management system (QMS) and products to meeting the new regulations. Now, nearly a year has passed since the amendment of the regulation. What is the status-quo of European medical device industry in terms of compliance with the new regulation?
The June 2023 survey on notified bodies indicates limited progress in the submitted applications and certificates issued. In comparison to the over 24’000 certificates issued under MDD and AIMDD, only around 13’000 MDR applications have been submitted and 3’900 certificates have been issued, representing only 16% certificates expected under the MDR. The situation with IVDs is even more concerning, with only 3% certificates issued for IVDs under the IVDR.
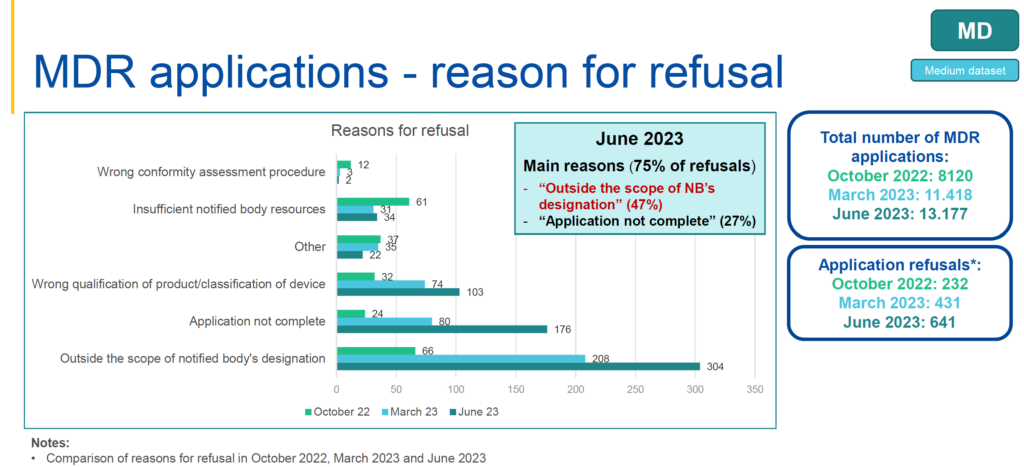
Unlike previous years, the availability of notified bodies is no longer a major reason for refusal of the application. To date, 40 and 12 notified bodies are designated under the MDR and IVDR, respectively. While, the capacity of notified bodies has gradually increased, the MDR and IVDR applications rate remains low. Concerning submitted applications, there is about 5% refusal rate for MDR and a 1% refusal rate for IVDR applications, respectively(see charts below). The top three reasons for MDR application refusals are:
- 47%: outside the scope of notified body’s designation;
- 27%: application isn’t complete;
- 16%: wrong qualification of product and classification of device.
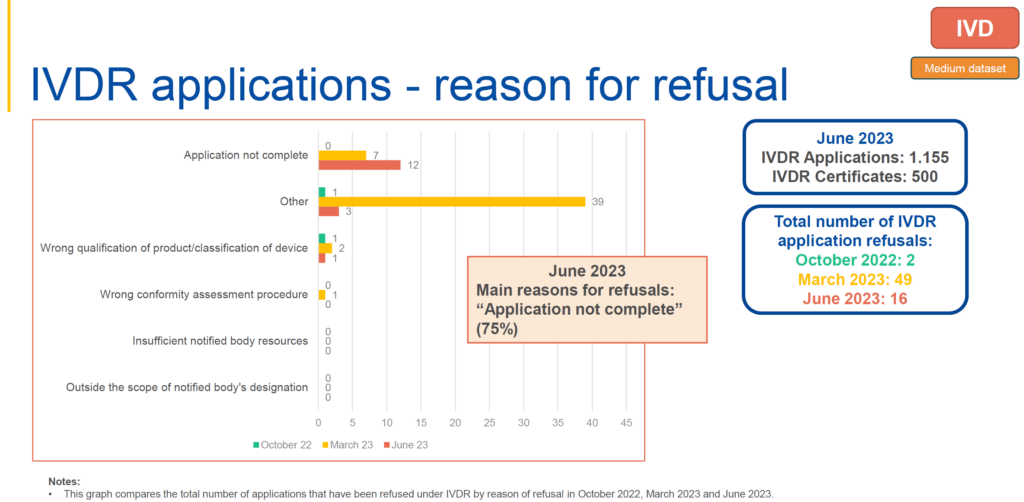
However, for IVDR applications, the major reason for refusal, accounting for 75%, is the incomplete nature of the applications, based on the relatively low number of IVDR applications submitted.
The 16% refusal rate of MDR applications due to wrong qualification and classification of device is indeed noteworthy. It suggests that for some manufacturers, navigating the classification of devices remains challenging, particularly given the broader scope of devices, technology innovation and the transition from MDD/IVDD to MDR/IVDR. This percentage appears relatively high and prompts the question of what manufacturers can and should do to prevent applications refusals stemming from incorrect classification of devices.
To mitigate the risk of classification-related refusals, manufacturers can take the following steps:
1. Thoroughly understand regulatory requirements:
- Ensure a comprehensive understanding of the MDR and IVDR classification rules (Annex VIII).
- Familiarize oneself with the definitions and requirements outlined in relevant guidance documents, particularly concerning borderline products that are general governed by Article 4 Regulatory status of products of the MDR and the corresponding Article 3 of the IVDR.
PS. borderline products are considered to be those cases where it is not clear from the outset whether a given product falls under MDR, IVDR or other regulations for medicinal products, biocides, cosmetic products, food, personal protective equipment or general consumer products.
2. Establish robust internal processes:
- Develop and implement a well-defined internal process for device classification, aligned with the MDR and IVDR requirements.
- Consider referring to key guidance documents, such as (MDCG 2021-24 and MDCG 2020-16, to establish standard operating procedures.
3. Utilize MDCG guidance documents:
- Leverage guidance documents provided by the Medical Device Coordination Group (MDCG), such as MDCG 2022-5 on the borderline between medical devices and medicinal products under the MDR. This guidance provides general discussion of the borderline between medical devices and medicinal products, including relevant definitions and examples.
- Stay updated on additional guidance, particularly the manual for borderline products. This manual contains all borderline cases submitted for consultation among competent authorities and finalised under the Helsinki Procedure. This manual is regularly updated by the Borderline and Classification Working Group (BCWG) which is a sub-group of MDCG.
4. Seek consultation when uncertain:
- When faced with uncertainty, especially in cases of new products, consider seeking consultation to clarify whether the product qualifies as a medical device.
- The manual for borderline products, regularly updated by the Borderline and Classification Working Group, can provide valuable insights and examples.
1 thought on “Classification remains challenging”
Comments are closed.