当今时代,生物科学技术是一个不断发展的行业,同时也受到政府主管部门高度监管。一个新产品,无论是药品还是创新型的医疗器械,从早期的概念性构想到最终的商业化,都需要经历多年的的研发,经历多个阶段性的进步。从早期的研发阶段(PH. Development),到产品注册过程(PH. REG)到后期的产品上市阶段(PH. Post market),都需要符合不断变化的新法规要求。我们提供专业的药品及医疗器械产品法律法规咨询,并且能够把这些法规要求具体转化在您日常的操作流程中。
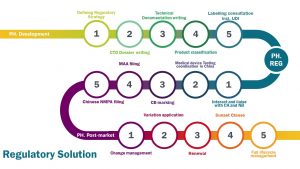
当今时代,生物科学技术是一个不断发展的行业,同时也受到政府主管部门高度监管。一个新产品,无论是药品还是创新型的医疗器械,从早期的概念性构想到最终的商业化,都需要经历多年的的研发,经历多个阶段性的进步。从早期的研发阶段(PH. Development),到产品注册过程(PH. REG)到后期的产品上市阶段(PH. Post market),都需要符合不断变化的新法规要求。我们提供专业的药品及医疗器械产品法律法规咨询,并且能够把这些法规要求具体转化在您日常的操作流程中。