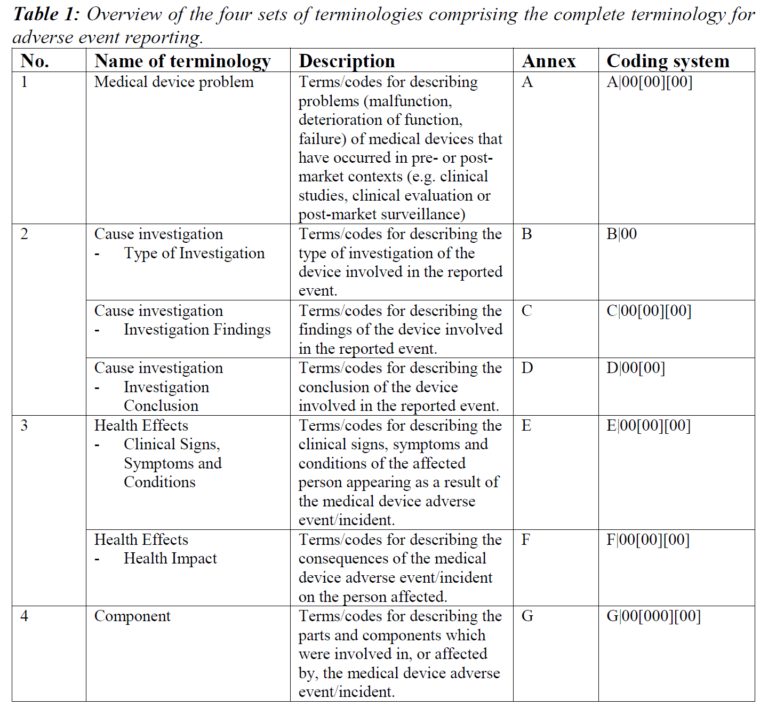
In practice, if a customer complains that a defibrillator failed to deliver a shock, this complaint can quickly be categorized as an adverse event according to its definition. This is because the failure to administer a shock to a patient with ventricular arrhythmia involving both faulty device and patients with clinical signs. To utilize the adverse event coding system, the manufacturer must determine the appropriate A-, B-, C-, D-, E-, F- and/or G-code sequentially at each step of its investigation. Below is a practical example of all adverse event coded identified by consulting the IMDRF guidance annexes.
- A071301 describes the medical device problem associated with the device’s failure to deliver electrical energy.
- B01 describes the type of investigation– testing of actual device was conducted to establish its functional and other properties.
- C0404 indicates that the investigational findings showed the adverse event was due to unintended compatibility.
- D09 is the investigational conclusion indicating that the problem was caused by inadequate information in the instruction for use.
- E060110 describes the clinical signs of the patient involved, which was ventricular arrhythmia.
- F05 describes the resulting consequences of the medical device adverse event on the patient, which was the delayed treatment.
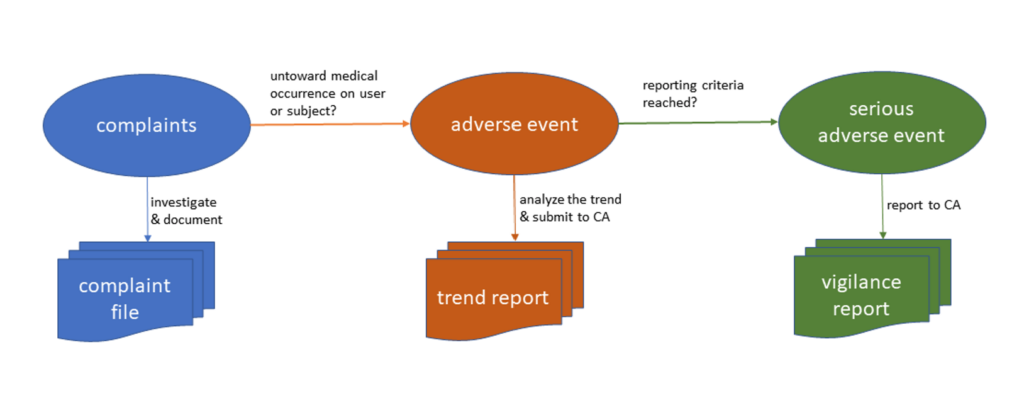
This AE meets the vigilance reporting criteria and thus requires a vigilance report to be submitted to the CA. When the manufacturer files the vigilance report (MIR template), all these A- to F-codes are mandatory for completing the report.
For non-SAEs it is also recommended to code AEs. When manufacturers consistently code all adverse events over time, they can systematically collect and analyze this data to determine if there is a significant increase compared to the foreseeable frequency or severity of AEs for the device, or category or group of devices over a specific period. If a trend is observed, manufacturers are required to submit a trend report to the CA to fulfill their regulatory requirements. Additionally, such systematic review of safety information and conclusion can feed back into risk management, potentially triggering a device redesign or improvement.